Drugs targeting a protein with a key role in neurodegeneration may slow the advance of the neurological disorder ALS Published online 31 May 2018
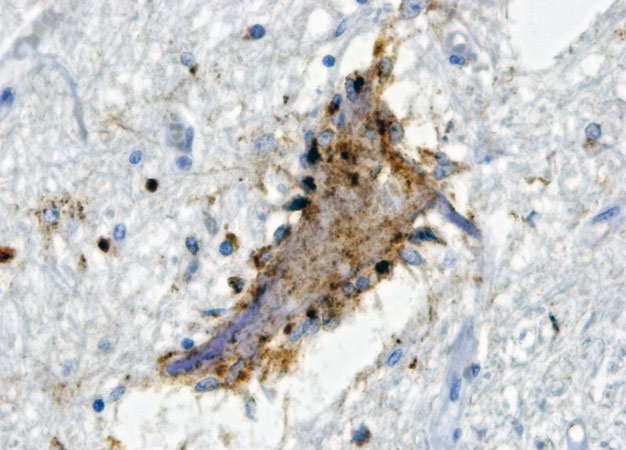
Motor neurons from a patient with ALS, where osteopontin is shown in gray. Blue indicates brain glial cells attacking the neuron.
© 2018 Hidemi Misawa A molecular pathway that becomes active in vulnerable subpopulations of neurons in people with the debilitating neurological disease amyotrophic lateral sclerosis (ALS) has been uncovered by Keio University researchers1. Therapies targeting this pathway could slow the progress of this incurable condition. The late cosmologist Stephen Hawking was the best known ALS sufferer. Hawking lived for 55 years after diagnosis, but about half of ALS patients die within three years of onset as the steady loss of motor neurons gradually robs sufferers of control over their bodies. Different types of motor neurons control specific kinds of muscle movement, and some are more likely to succumb to ALS than others: motor neurons controlling slow-twitch muscles are largely resistant to degeneration, whereas those controlling fast-twitch muscles are susceptible. There is additional complexity ― a subset of these latter motor neurons perishes early in ALS, while another survives until its second wave, during which patients lose most voluntary movements. "The different vulnerabilities of different types of motor neurons is one of the long-standing mysteries for ALS researchers," notes Hidemi Misawa at Keio University's Faculty of Pharmacy. This has led to a search for biological markers that clarify the nature of this susceptibility and enable early discrimination of endangered motor neurons. Misawa's team recently obtained evidence that the cell signaling protein osteopontin might be a key to understanding the selective disease process of ALS. "It's expressed abundantly in motor neurons serving ALS-resistant muscles," Misawa says. The researchers have now determined that this protein plays a key role in establishing the tipping point of vulnerability to ALS, and they have identified osteopontin-activated molecular pathways that indicate which cells will go from being protected to becoming vulnerable to degeneration. To assess this protein's function, the researchers knocked out the gene that encodes it in a mouse model of ALS. Osteopontin deficiency initially delayed the onset of neurodegeneration and muscle loss in these animals, but the disease progressed faster than usual once it began to manifest. This reflects osteopontin's dual role. On the one hand, it stimulates the induction and activation of the enzyme matrix metalloproteinase-9 (MMP-9), which renders neurons susceptible to degeneration. This explains why osteopontin deficiency initially delays ALS. On the other hand, osteopontin sends signals that protect against inflammatory damage from brain glial cells, so that its absence ultimately leaves neurons vulnerable to destruction. These findings suggest that osteopontin could be a potential drug target for ALS therapy. Misawa hypothesizes that blocking osteopontin's activation of MMP-9 in early-stage ALS patients could slow disease progression. His team is now using more-sophisticated mouse models to study this pathway in greater detail.About the researcher

Hidemi Misawa― Professor
Department of Pharmaceutical Sciences, Division of Pharmacology
Hidemi Misawa is a professor at Keio University's Division of Pharmacology and the director of the Research Center for Drug Discovery at the university's Faculty of Pharmacy. His research interests include molecular mechanisms of neurodegenerative diseases, cholinergic control of higher brain functions, and neuroimmune control by vagal neurons.
Links
Reference
- Morisaki, Y., Niikura, M., Watanabe, M., Onishi, K., Tanabe, S., Moriwaki, Y. et al. Selective expression of osteopontin in ALS-resistant motor neurons is a critical determinant of late phase neurodegeneration mediated by matrix metalloproteinase-9. Scientific Reports 6, 27354 (2016). | article